有机发光
Singlet State vs Triplet State
Understanding the difference between fluorescence and phosphorescence requires the knowledge of electron spin and the differences between singlet and triplet states. The Pauli Exclusion principle states that two electrons in an atom cannot have the same four quantum numbers \((n, l, m_{l}, m_{s})\) and only two electrons can occupy each orbital where they must have opposite spin states. These opposite spin states are called spin pairing. Because of this spin pairing, most molecules do not exhibit a magnetic field and are diamagnetic(抗磁的). In diamagnetic molecules, electrons are not attracted or repelled by the static electric field. Free radicals are paramagnetic(顺磁的) because they contain unpaired electrons have magnetic moments that are attracted to the magnetic field.
Singlet state is defined when all the electron spins are paired in the molecular electronic state and the electronic energy levels do not split when the molecule is exposed into a magnetic field. A doublet state occurs when there is an unpaired electron that gives two possible orientations when exposed in a magnetic field and imparts different energy to the system. A singlet or a triplet can form when one electron is excited to a higher energy level. In an excited singlet state, the electron is promoted in the same spin orientation as it was in the ground state (paired). In a triplet excited stated, the electron that is promoted has the same spin orientation (parallel) to the other unpaired electron.
Singlet, doublet and triplet is derived using the equation for multiplicity \( 2S+1\), where \(S\) is the total spin angular momentum (sum of all the electron spins).
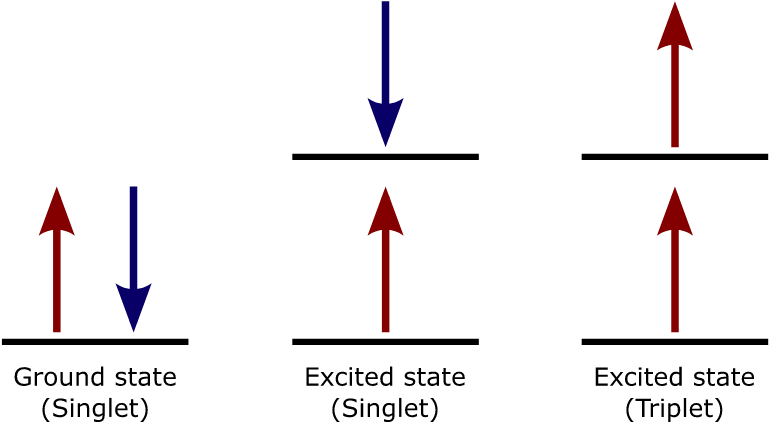
上图是Spin in the ground and excited states,处在ground state(singlet)的时候是diamagnetic,处在excited state(triplet)的时候是paramagnetic。This difference in spin state makes the transition from singlet to triplet (or triplet to singlet) more improbable than the singlet-to-singlet transitions. This singlet to triplet (or reverse) transition involves a change in electronic state. For this reason, the lifetime of the triplet state is longer the singlet state by approximately \(10^4\) seconds fold difference. The radiation that induced the transition from ground to excited triplet state has a low probability of occurring, thus their absorption bands are less intense than singlet-singlet state absorption. The excited triplet state can be populated from the excited singlet state of certain molecules which results in phosphorescence. These spin multiplicities in ground and excited states can be used to explain transition in photoluminescence molecules by the Jablonski diagram.
Jablonski Diagrams
The Jablonski diagram that drawn below is a partial energy diagram that represents the energy of photoluminescent molecule in its different energy states. The lowest and darkest horizontal line represents the ground-state electronic energy of the molecule which is the singlet state labeled as \(S_0\). At room temperature, majority of the molecules in a solution are in this state.
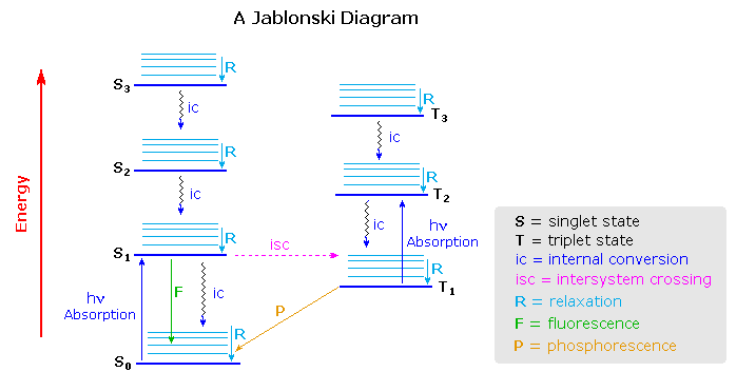
The upper darkest line represents the ground vibrational state of the three excited electronic state. The energy of the triplet state is lower than the energy of the corresponding singlet state. (和洪德最大多重度规则有关吗?)There are numerous vibrational levels that can be associated with each electronic state as denoted by the thinner lines. Singlet到Triplet的transition leads to a change in multiplicity and thus has a low probability of occurring which is a forbidden transition. Molecules also go through vibration relaxation to lose any excess vibrational energy that remains when excited to the electronic states ( \(S_1\) and \(S_2\) ) as demonstrated in wavy lines. The knowledge of forbidden transition is used to explain and compare the peaks of absorption and emission.
Relaxation and Fluorescence
When an electron is promoted to an electronic excited state, it often ends up in an excited vibrational state as well. Thus, some of the energy put into electronic excitation is immediately passed into vibrational energy. Vibrational energy, however, doesn't just travel in photons. It can be gained or lost through molecular collisions and heat transfer.
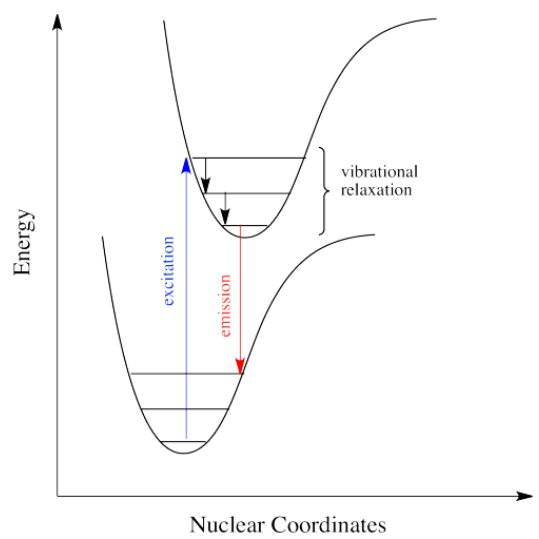
Just how does a molecule undergo vibrational relaxation? Vibrational energy is the energy used to lengthen or shorten bonds, or to widen or squeeze bond angles.
(1) 大分子:Given a big enough molecule, some of this vibrational energy could be transferred into bond lengths and angles further away from the electronic transition.
(2) 小分子:Otherwise, if the molecule is small, it may transfer some of its energy in collisions with other molecules.
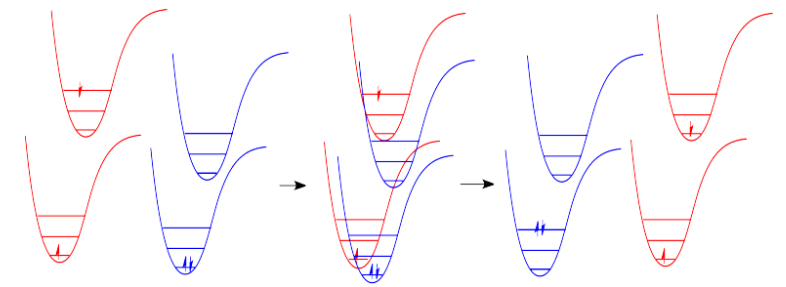
In molecules, as one molecule drops to a lower vibrational state, the other will hop up to a higher vibrational state with the energy it gains. In the drawing below, the red molecule is in an electronic excited and vibrational state. In a collision, it transfers some of its vibrational energy to the blue molecule.
Radiationless Transitions: Internal Conversion
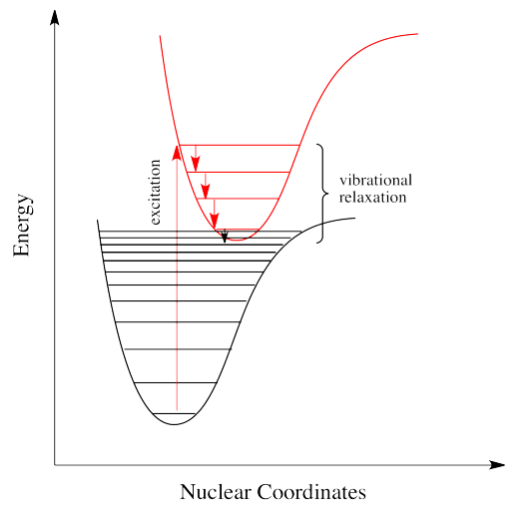
If electrons can get to a lower energy state, and give off a little energy at a time, by hopping down to lower and lower vibrational levels, do they need to give off a giant photon at all? Maybe they can relax all the way down to the ground state via vibrational relaxation. That is certainly the case. Given lots of vibrational energy levels, and an excited state that is low enough in energy so that some of its lower vibrational levels overlap with some of the higher vibrational levels of the ground state, the electron can hop over from one state to the other, without releasing a photon.
This event is called a "radiationless transition", because it occurs without release of a photon. The electron simply slides over from a low vibrational state of the excited electronic state to a high vibrational state of the electronic ground state. If the electron simply keeps dropping a vibrational level at a time back to the ground state, the process is called "internal conversion".
Internal conversion has an important consequence. Because the absorption of UV and visible light can result in energy transfer into vibrational states, much of the energy that is absorbed from these sources is converted into heat. That can be a good thing if you happen to be a marine iguana trying to warm up in the sun after a plunge in the icy Pacific. It can also be a tricky thing if you are a process chemist trying to scale up a photochemical reaction for commercial production of a pharmaceutical, because you have to make sure the system has adequate cooling available.
Radiationless Transitions: Intersystem Crossing
There is a very similar event, called 【intersystem crossing】, that leads to the electron getting caught between the excited state and the ground state. Just as, little by little, vibrational relaxation can lead the electron back onto the ground state energy surface, it can also lead the electron into states that are intermediate in energy.
For example, suppose an organic molecule undergoes electronic excitation. Generally, organic molecules have no unpaired electrons. Their ground states are singlet states. According to one of our selection rules for electronic excitation, the excited state must also have no unpaired electrons. In other words, the spin on the electron that gets excited is the same after excitation as it was before excitation.
However, that's not the lowest possible energy state for that electron. When we think about atomic orbital filling, there is a rule that governs the spin on the electrons in degenerate orbitals: in the lowest energy state, spin is maximized (Hund's rule). In other words, when we draw a picture of the valence electron configuration of nitrogen, we show nitrogen's three p electrons each in its own orbital, with their spins parallel.
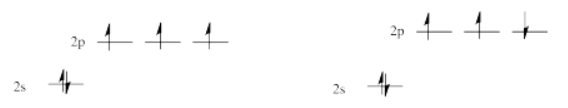
The picture with three unpaired electrons, all with parallel spins, shows a nitrogen in the quartet spin state. Having one of those spins point the other way would result in a different spin state. One pair of electrons in the \( p\) level would be spin-paired, one up and one down, even though they are in different \( p\) orbitals. That would leave one electron without an opposite partner. The nitrogen would be in a doublet spin state. That is not what happens. The spin state on the left is lower in energy than the state on the right. That's just one of the rules of quantum mechanics (Hund's rule): maximize spin when orbitals are singly occupied.
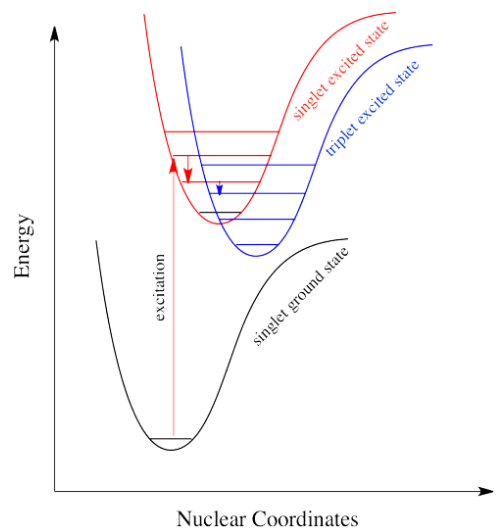
It's the same in a molecule with the triplet state lower in energy than the singlet state. Why didn't the electron get excited to the triplet state in the first place? That's against the rules. But sliding down vibrationally onto the triplet state from the singlet excited state is not, because it doesn't involve absorption of a photon.
 Intersystem crossing can have important consequences in reaction chemistry because it allows access to triplet states that are not normally avaiable in many molecules. Because triplet states feature unpaired electrons, their reactivity is often typified by radical processes. That means an added suite of reactions can be accessed via this process.
Intersystem crossing can have important consequences in reaction chemistry because it allows access to triplet states that are not normally avaiable in many molecules. Because triplet states feature unpaired electrons, their reactivity is often typified by radical processes. That means an added suite of reactions can be accessed via this process.
Intersystem crossing is one way a system can end up in a triplet excited state. Even though this state is lower in energy than a singlet excited state, it cannot be accessed directly via electronic excitation because that would violate the spin selection rule (\( \Delta S=0\)). That's where the electron gets stuck, though. The quick way back down to the bottom is by emitting a photon, but because that would involve a change in spin state, it is not allowed.(可通过spin-orbit coupling进行) Realistically speaking, that means it takes a long time. By "a long time", we might mean a few seconds, several minutes, or possibly even hours. Eventually, the electron can drop back down, accompanied by the emission of a photon. This situation is called "phosphorescence".
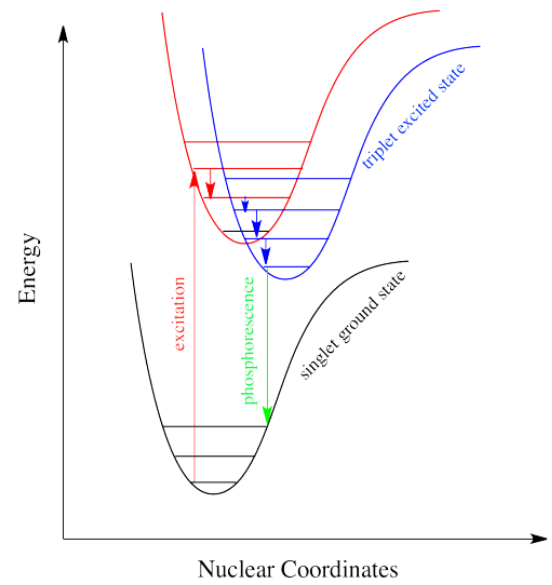
Molecules that display phosphorescence are often incorporated into toys and shirts so that they will glow in the dark.
注:除了内转换,还有【外转换】,激发分子与溶剂或其他分子之间产生相互作用而转移能量的非辐射跃迁。
Molecular Absorption of Light
When we were talking about the various sorts of molecular orbitals present in organic compounds earlier, you will have come across this diagram showing their relative energies:
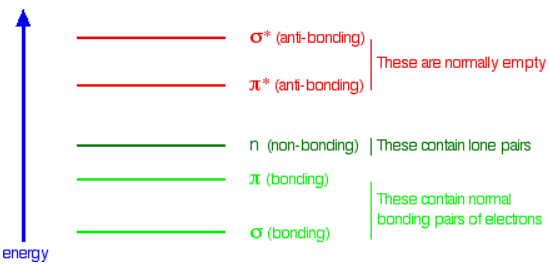 When light passes through the compound, energy from the light is used to promote an electron from a bonding or non-bonding orbital into one of the empty anti-bonding orbitals. The possible electron jumps that light might cause are:
When light passes through the compound, energy from the light is used to promote an electron from a bonding or non-bonding orbital into one of the empty anti-bonding orbitals. The possible electron jumps that light might cause are:
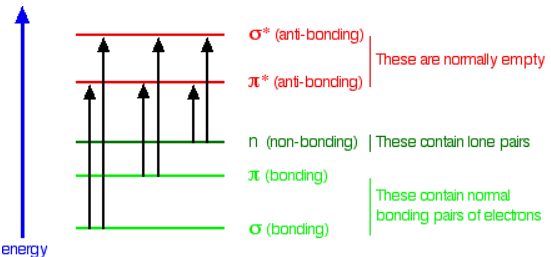 In each possible case, an electron is excited from a full orbital into an empty anti-bonding orbital. Each jump takes energy from the light, and a big jump obviously needs more energy than a small one. Each wavelength of light has a particular energy associated with it. If that particular amount of energy is just right for making one of these energy jumps, then that wavelength will be absorbed - its energy will have been used in promoting an electron.
In each possible case, an electron is excited from a full orbital into an empty anti-bonding orbital. Each jump takes energy from the light, and a big jump obviously needs more energy than a small one. Each wavelength of light has a particular energy associated with it. If that particular amount of energy is just right for making one of these energy jumps, then that wavelength will be absorbed - its energy will have been used in promoting an electron.
An absorption spectrometer works in a range from about 200 nm (in the near ultra-violet) to about 800 nm (in the very near infra-red). Only a limited number of the possible electron jumps absorb light in that region. Look again at the possible jumps. This time, the important jumps are shown in black, and a less important one in grey. The grey dotted arrows show jumps which absorb light outside the region of the spectrum we are working in.
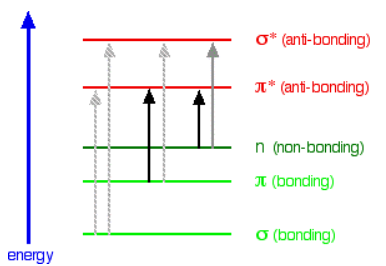 Remember that bigger jumps need more energy and so absorb light with a shorter wavelength. The jumps shown with grey dotted arrows absorb UV light of wavelength less that 200 nm. The important jumps are:
Remember that bigger jumps need more energy and so absorb light with a shorter wavelength. The jumps shown with grey dotted arrows absorb UV light of wavelength less that 200 nm. The important jumps are:
- from \(\pi \)bonding orbitals to \(\pi \)anti-bonding orbitals;
- from non-bonding orbitals to \(\pi \) anti-bonding orbitals;
- from non-bonding orbitals to sigma anti-bonding orbitals.
That means that in order to absorb light in the region from 200 - 800 nm (which is where the spectra are measured), the molecule must contain either \( \pi\) bonds or atoms with non-bonding orbitals. Remember that a non-bonding orbital is a lone pair on, say, oxygen, nitrogen or a halogen. Groups in a molecule which absorb light are known as chromophores.
The diagram below shows a simple UV-visible absorption spectrum for buta-1,3-diene - a molecule we will talk more about later. Absorbance (on the vertical axis) is just a measure of the amount of light absorbed. The higher the value, the more of a particular wavelength is being absorbed.
UV-visible Absorption spectrum
he diagram below shows a simple UV-visible absorption spectrum for buta-1,3-diene - a molecule we will talk more about later. Absorbance (on the vertical axis) is just a measure of the amount of light absorbed. The higher the value, the more of a particular wavelength is being absorbed.
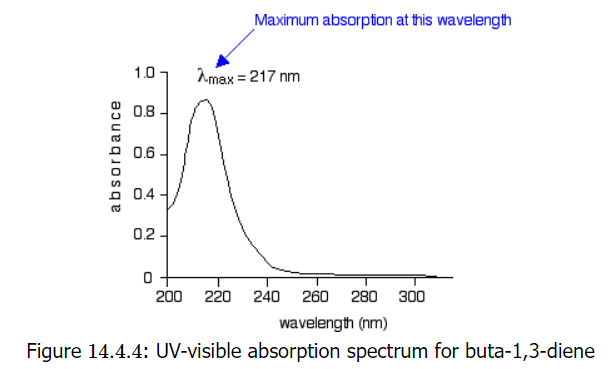
You will see that absorption peaks at a value of 217 nm. This is in the ultra-violet and so there would be no visible sign of any light being absorbed - buta-1,3-diene is colorless. In buta-1,3-diene, CH2=CH-CH=CH2, there are no non-bonding electrons. That means that the only electron jumps taking place (within the range that the spectrometer can measure) are from \( \pi\) bonding to \( \pi\) anti-bonding orbitals.
A chromophore such as the carbon-oxygen double bond in ethanal, for example, obviously has \( \pi\) electrons as a part of the double bond, but also has lone pairs on the oxygen atom. That means that both of the important absorptions from the last energy diagram are possible. You can get an electron excited from a \( \pi\) bonding to a \( \pi\) anti-bonding orbital, or you can get one excited from an oxygen lone pair (a non-bonding orbital) into a \( \pi\) anti-bonding orbital.
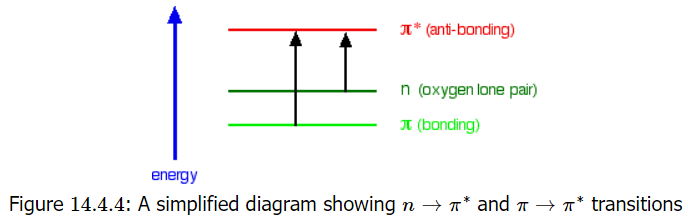 The non-bonding orbital has a higher energy than a \( \pi\) bonding orbital. That means that the jump from an oxygen lone pair into a \( \pi\) anti-bonding orbital needs less energy. That means it absorbs light of a lower frequency and therefore a higher wavelength. For example, ethanal can therefore absorb light of two different wavelengths:
The non-bonding orbital has a higher energy than a \( \pi\) bonding orbital. That means that the jump from an oxygen lone pair into a \( \pi\) anti-bonding orbital needs less energy. That means it absorbs light of a lower frequency and therefore a higher wavelength. For example, ethanal can therefore absorb light of two different wavelengths:
- the \( \pi\) bonding to \( \pi\) anti-bonding absorption peaks at 180 nm. These \(n \rightarrow \pi^{*}\) transitions involve moving an electron from a nonbonding electron pair to a antibonding \( \pi^{*}\) orbital. They tend to have molar absorbtivities less than 2000;
- the non-bonding to \( \pi\) anti-bonding absorption peaks at 290 nm. These \( \pi \rightarrow \pi^{*}\) transitions involve moving an electron from a bonding \(\pi^{*}\) orbital to an antibonding \(\pi^{*}\) orbital. They tend to have molar absorptivities on the order of 10,000.
- 上面好像写反了。
Conjugation and Delocalization
Consider these three molecules: Ethene contains a simple isolated carbon-carbon double bond, but the other two have conjugated double bonds. In these cases, there is delocalization of the \( \pi\) bonding orbitals over the whole molecule. Now look at the wavelengths of the light which each of these molecules absorbs.
Ethene contains a simple isolated carbon-carbon double bond, but the other two have conjugated double bonds. In these cases, there is delocalization of the \( \pi\) bonding orbitals over the whole molecule. Now look at the wavelengths of the light which each of these molecules absorbs.
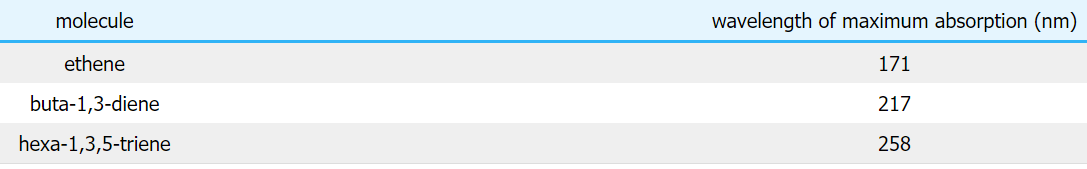
All of the molecules give similar UV-visible absorption spectra - the only difference being that the absorptions move to longer and longer wavelengths as the amount of delocalization in the molecule increases. Why is this? You can actually work out what must be happening.
- The maximum absorption is moving to longer wavelengths as the amount of delocalization increases.
- Therefore maximum absorption is moving to shorter frequencies as the amount of delocalization increases.
- Therefore absorption needs less energy as the amount of delocalization increases.
- Therefore there must be less energy gap between the bonding and anti-bonding orbitals as the amount of delocalization increases.
Compare ethene with buta-1,3-diene. In ethene, there is one \( \pi\) bonding orbital and one \( \pi\) anti-bonding orbital. In buta-1,3-diene, there are two \( \pi\) bonding orbitals and two \( \pi\) anti-bonding orbitals. This is all discussed in detail on the introductory page that you should have read.
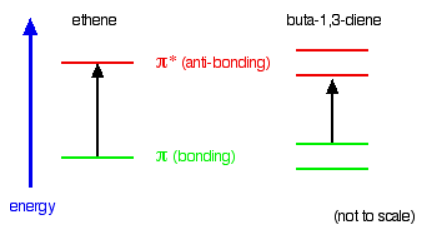
The highest occupied molecular orbital is often referred to as the HOMO - in these cases, it is a \( \pi\) bonding orbital. The lowest unoccupied molecular orbital (the LUMO) is a \( \pi\) anti-bonding orbital. Notice that the gap between these has fallen. It takes less energy to excite an electron in the buta-1,3-diene case than with ethene. In the hexa-1,3,5-triene case, it is less still.
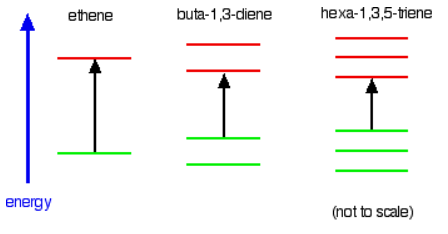 If you extend this to compounds with really massive delocalization, the wavelength absorbed will eventually be high enough to be in the visible region of the spectrum, and the compound will then be seen as colored. A good example of this is the orange plant pigment, beta-carotene - present in carrots, for example.
If you extend this to compounds with really massive delocalization, the wavelength absorbed will eventually be high enough to be in the visible region of the spectrum, and the compound will then be seen as colored. A good example of this is the orange plant pigment, beta-carotene - present in carrots, for example.
(1) Why is beta-carotene orange?
Beta-carotene has the sort of delocalization that we've just been looking at, but on a much greater scale with 11 carbon-carbon double bonds conjugated together. The diagram shows the structure of beta-carotene with the alternating double and single bonds shown in red.The more delocalization there is, the smaller the gap between the highest energy \( \pi\) bonding orbital and the lowest energy \( \pi\) anti-bonding orbital. To promote an electron therefore takes less energy in beta-carotene than in the cases we've looked at so far - because the gap between the levels is less. Beta-carotene absorbs throughout the ultra-violet region into the violet - but particularly strongly in the visible region between about 400 and 500 nm with a peak about 470 nm.
Color Wheel
(2) CB/VB 和LUMO/HOMO对比[Thomas Jüstel-PPT]
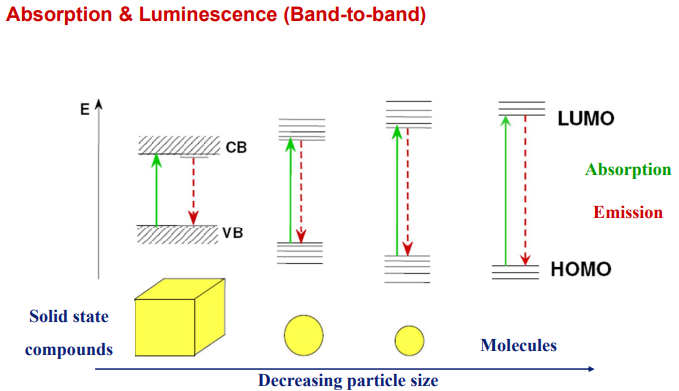
Franck-Condon: Electronic and Vibrational Coupling
So far, we have come across one big rule of photon absorbance. To be absorbed, a photon's energy has to match an energy difference within the compound that is absorbing it. Different wavelengths would be able to promote different electrons, depending on the energy difference between an occupied electronic energy level and an unoccupied one. Other types of electromagnetic radiation would not be able to promote an electron, but they would be coupled to other events. For example, absorption of infrared light is tied to vibrational energy levels. Microwave radiation is tied to rotational energy levels in molecules. Thus, one reason a photon may or may not be absorbed has to do with whether its energy corresponds to the available energy differences within the molecule or ion that it encounters.
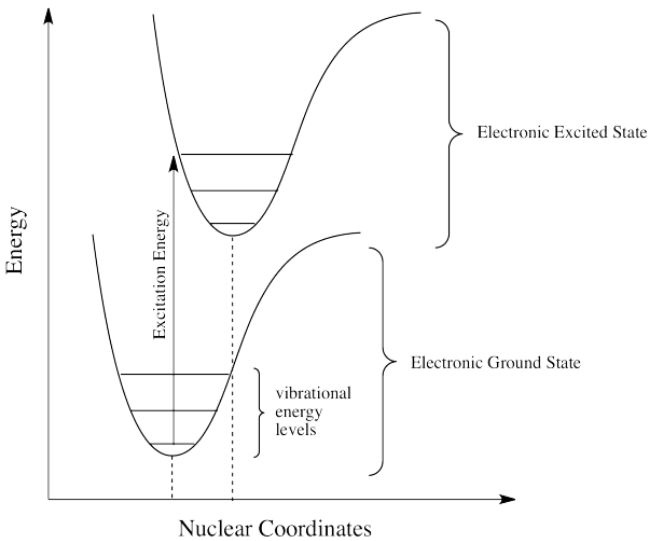
Photons face other limitations. One of these is a moderate variation on our main rule. It is called the Frank Condon Principle. According to this idea, when an electron is excited from its normal position, the ground state, to a higher energy level, the optimal positions of atoms in the molecule may need to shift. Because electronic motion is much faster than nuclear motion, however, any shifting of atoms needed to optimize positions as they should be in the excited state will have to wait until after the electron gets excited. In that case, when the electron lands and the atoms aren't yet in their lowest energy positions for the excited state, the molecule will find itself in an excited vibrational state as well as an excited electronic state. That means the required energy for excitation does not just correspond to the difference in electronic energy levels; it is fine-tuned to reach a vibrational energy level, which is quantized as well.
影响荧光的主要因素
(1) 荧光助色团与荧光消色团
可使化合物荧光增强的基团被称为荧光助色团。一般为给电子取代基, 如-NH2、-OH等。相反, 吸电子基团如 -COOH 、-CN 等将减弱或抑制荧光的产生,被称为荧光消色团。
(2) 增加稠合环可增强荧光
增加共平面的稠合环的数目, 特别是当稠合环以线型排列时,将有利于体系内\(\pi\)电子的流动, 从而使体系发生跃迁所需吸收的能量降低, 进而有利于荧光的产生。
(3) 提高分子的刚性可增强荧光
刚性增强后,将减弱分子的振动,从而使分子的激发能不易因振动而以热能形式释放;另外,分子刚性的增加常有利于增加分子的共平面,从而有利于增大分子内\(\pi\)电子的流动性,也就有利于荧光的产生。
(4) 激发态电子组态的影响
根据电子跃迁规则,\(S_{1}\)态的电子组态是\(\left(\pi, \pi^{*}\right)\)态时,有利于荧光的产生, 当\(\mathrm{S}_{1}\)态的电子组态是\(\left(n, \pi^{*}\right)\)态时, 不利于荧光的产生。
(5) 重原子将导致荧光量子产率的降低
重原子具有增强系间窜越的作用,将增大从\(\mathrm{S}_{1}\)态向\(\mathrm{T}_{1}\)态的系间窜越的速率常数和量子产率,从而导致降低荧光量子产率。
(6) 溶剂极性的影响
降低体系温度可以提高苂光量子产率。
(7) 其它影响因素
如氢键、吸附、溶剂粘度增加等均可提高荧光的量子产率,这都可用减少了分子的热振动和增加了分子的刚性来解释。
有机长余辉发光
Lanthanide Complexes
TR-FRET用稀土螯合物
镧系元素螯合物的独特性质: 强荧光性组的Eu3+、Sm3+、Tb3+、Dy3+,通常在水溶液中只能发射极为微弱的 荧光,普通荧光光度计无法检出。但当它们形成适当的配体和 络合物(以下称为镧系元素螯合物)时,可发射出强荧光。下 面将具体介绍这个过程。经近紫外范围的光照射后,配体首先 吸收光能量而受到激发。然后,络合物通过能量转移从单重态 跃迁到三重态*2,之后能量转移至位于中心的镧系元素离子 并使之转化为激发态。随后,镧系元素离子从激发态跃迁回基 态,发出荧光(发射光的辐射迁移)。由于镧系元素离子的受 激能级与基态能级之间的能量差较大,因此出现非辐射迁移的 几率较小,荧光量子产率较高。镧系元素螯合物与荧光素等有 机荧光染料相比,具有以下几点突出的荧光特性。
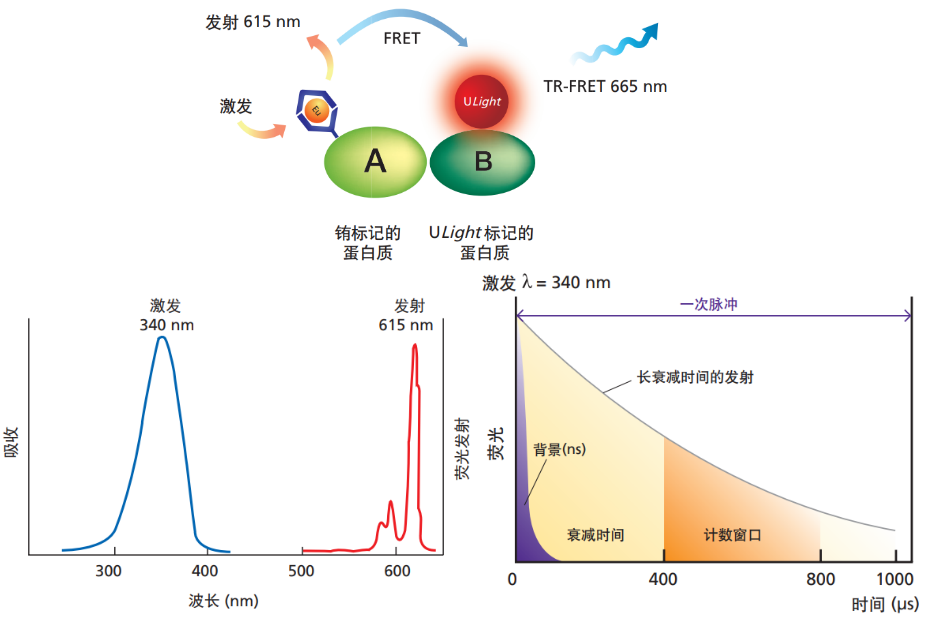
(1) 荧光寿命增加了100,000 倍
镧系元素离子以荧光寿命长著称。有机荧光染料的荧光寿命通常为纳秒级,然而以铕螯合物为例,其荧光寿命可达到1,000微秒以上。铕螯合物的荧光寿命非常长,FRET过程中受体的荧光寿命也较长。如此一来,当采用时间分辨荧光检测进行测定时,就可以避免样品中的杂质以及样品板材料等生成的短寿命背景荧光。
(2) 较大的斯托克斯位移
有机荧光染料的斯托克斯位移为数十纳米,而且其激发后荧光 光谱通常存在较大的重叠。然而以铕螯合物为例,其斯托克斯 位移非常大,可达到约300nm。这是因为激发和发射 发生在络合物的不同部位,激发光谱取决于配体,而荧光光谱 取决于位于中心的Eu3+(与配体无关)。这样就可避免激发 光以及激发光引起的散射光产生干扰。
(3) 荧光峰的峰形尖锐
以铕螯合物为例,615nm辐射的荧光能量主要集中在615±10nm波长范围内。因此,虽然荧光量子产率较低,但与有机荧光染料的荧光光谱相比,特定波长处的荧光强度仍大得多。有机荧光染料会发生的浓度猝灭*3现象也基本不会出现。目前为止,人们已合成了大量用于时间分辨荧光检测和TR-FRET检测的配体,现在,使用这类配体的各种各样的系统得到了广泛应用。
使用铕螯合物的时间分辨荧光检测:时间分辨荧光检测利用物质荧光寿命的差异来选择和检测具有长荧光寿命的目标分子。在一般的荧光检测中,样品受到激发光照射时,其中的杂质和样品容器材料也会受到激发而发出荧光,这些自体荧光和激发光的散射光等会与目标荧光一起被检测到。然而,这类荧光的寿命为10纳秒左右,很快就会衰减。使用荧光寿命较长的铕螯合物,等到背景荧光已完全衰减后再 检测荧光(时间分辨荧光检测),就能够以较高的灵敏度单一 地检测铕螯合物的荧光。此外,由于铕螯合物具有较 大的斯托克斯位移,因此可获得较高的S/B。使用具备时间分辨检测功能的多级计数器将340nm的激发光按每秒1,000次脉冲的频率进行照射。每一次脉冲期间,等待400微秒后,测量400至800微秒期间的荧光。
聚集诱导发光(AIE)
参考资料:
(1) 【材料】J. Phys. Chem. Lett.┃聚集诱导发光的机理?
(2) 唐本忠院士团队:聚集诱导发光的基本理解及未来发展
(3) 站在巨人的肩膀上,中国科学家看到了更高更远的地方(科技袁人)
磷光(Phosphorescence)$$\mathrm{S}_{0}+h \nu \rightarrow \mathrm{S}_{1} \rightarrow \mathrm{T}_{1} \rightarrow \mathrm{S}_{0}+h \nu^{\prime}$$其中\( \mathrm{S}\)是单重态,\(\mathrm{T}\)是三重态,其下标表示状态(\(0\)是基态,\( 1\)是激发态)。跃迁也可以发生在更高的激发态,但这里简单起见只用了第一激发态表示。
其中 S 是单重态,T 是三重态,其下标表示状态(0 是基态,1 是激发态)。跃迁也可以发生到更高的能级,但为简单起见,表示第一个激发态。
OLED
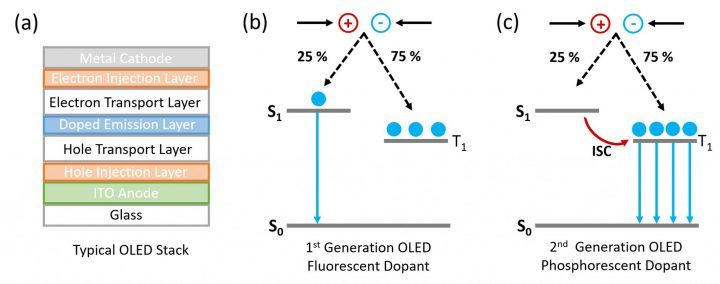
Fluorescence: S(单线态)到S的跃迁;
Phosphorescence: T(单线态)到S的跃迁;
概述:OLEDs are one of the most popular display types for televisions and smartphones, offering higher contrast ratios and lower power consumption than conventional LCD displays. In an OLED, layers of organic (carbon-based) semiconductors are sandwiched between two electrodes and electrons and holes are injected into the organic semiconductor under an applied bias. Upon encountering, the electrons and holes first form Coulombically bound electron-hole pairs called excitons which can then recombine to generate light. Due to spin statistics, 25% of the excitons formed will be in the singlet state (S1) and 75% in the triplet state (T1).
第一代基于Fluorescence发光的OLED:使用的是fluorescent molecular emitters。OLED的发光效率将取决于两种自旋状态产生的百分比,而很遗憾的是,singlets占有的比例仅有25%,这也就决定了,最终至多只能有25%的能量转化为光线。
第二代基于Phosphorence发光的OLED:To overcome this limitation; heavy metals, such as platinum and iridium, were incorporated into the molecular emitters to make 2nd generation phosphorescent OLEDs. The presence of heavy metals in the molecule increases the strength of the spin-orbit coupling between the spin angular momentum and the orbital angular momentum and the T1 > S0 radiative transition becomes allowed. This approach enables IQEs of 100% to be achieved; however, the use of heavy metals has several significant drawbacks. The metals are rare and expensive and are therefore impracticable for high production volumes. In addition, phosphorescent OLEDs suffer from poor stability, particularly in the blue, and to date, no stable deep blue phosphorescent emitter has been found.
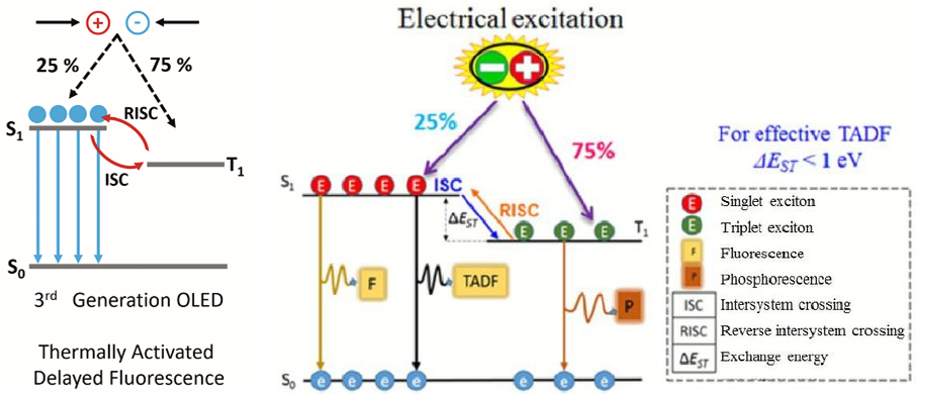
第三代基于Phosphorence发光的OLED:These shortcomings led to the development of heavy metal free 3rd generation OLEDs which operate using the TADF mechanism. In a TADF emitter, the S1 and T1 states are designed to be close in energy and strongly coupled, which enables excitons generated the T1 state to undergo a thermally assisted reverse intersystem crossing (RISC) to the S1 state where they can then radiatively decay to the S0 resulting in delayed fluorescence emission. Using TADF, IQEs of 100% can be achieved without the need for heavy metals. New TADF emitters with high quantum yields, good stability and desirable colour coordinates need to be developed.
注:TADF表示Thermally activated delayed fluorescence
参考资料:
(1) Edinburgh Instruments-OLED
(2) OLED的制造工艺及关键技术概述-中科院半导体所
(2) 同一材料的电致发光和光致发光光谱相同吗?-知乎
(3) 漫谈照明技术历史、发展和科学原理-蔻享
(4) Nature:世界首次!攻克荧光极限!(拓展)